发布时间:2023-09-05
基于超高分辨率液质联用质谱的定量蛋白质组学已成为重要的生命科学研究手段。与其它组学方法一样,恰当的数据分析流程是蛋白质组学研究的关键。基于质谱的蛋白质组数据具有方法仪器多样、搜库定量软件繁多的特点,导致下游的数据处理分析更为困难繁琐,亟需一个可高效处理多样的蛋白质组学数据的下游分析工具。
近日,中国科学院广州生物医药与健康研究院张小飞团队在Bioinformatics发表题为DEP2: an upgraded comprehensive analysis toolkit for quantitative proteomics data的论文。该成果揭示了一个可用于处理多种蛋白质组定量软件输入数据的R包DEP2。DEP2是一个用于定量蛋白质组学数据下游处理、差异分析、可视化的工具R包,利用S4对象进行数据打包并通过limma进行差异分析。DEP2集成肽段至蛋白重定量算法、缺失值填充方法,可用于分析肽段层面和蛋白组层面的定量蛋白质组、修饰蛋白质组数据。同时DEP2捆绑了一个用模块化搭建的shiny应用,可以无代码方式同时处理多个蛋白质组学数据。DEP2兼容多类型上游结果,为研究人员处理蛋白质组数据提供便利。该R包已开源发布在https://github.com/mildpiggy/DEP2。
广州健康院博士研究生冯振桓、新乡医学院范培杨为该论文共同第一作者。广州健康院张小飞研究员为该论文通讯作者。该研究成果得到了国家重点研发计划、广东省科技计划项目、香港创新科技署项目的资助。
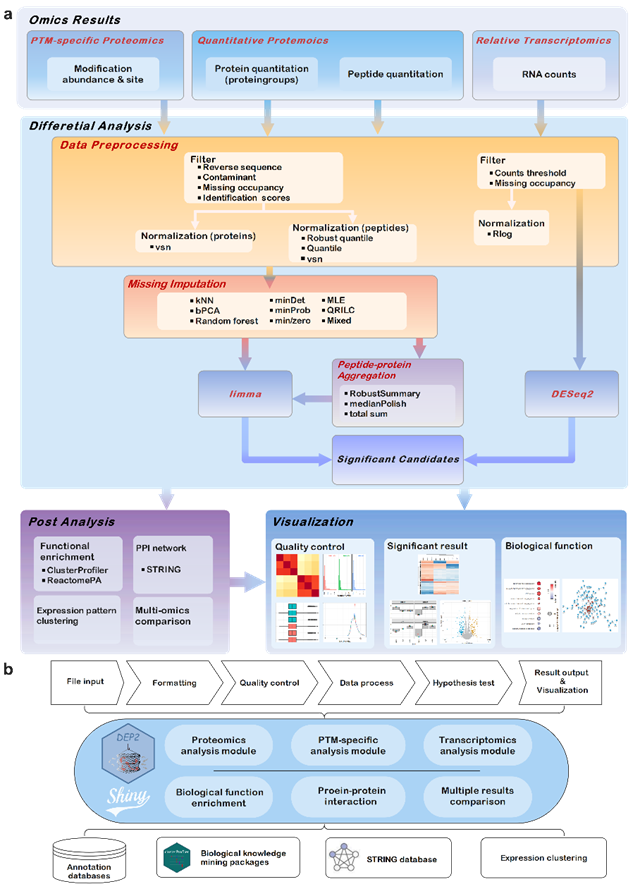
DEP2分析流程和内置Shiny APP 示意图
附件下载:





